Analysis Pipeline
FASTQ input
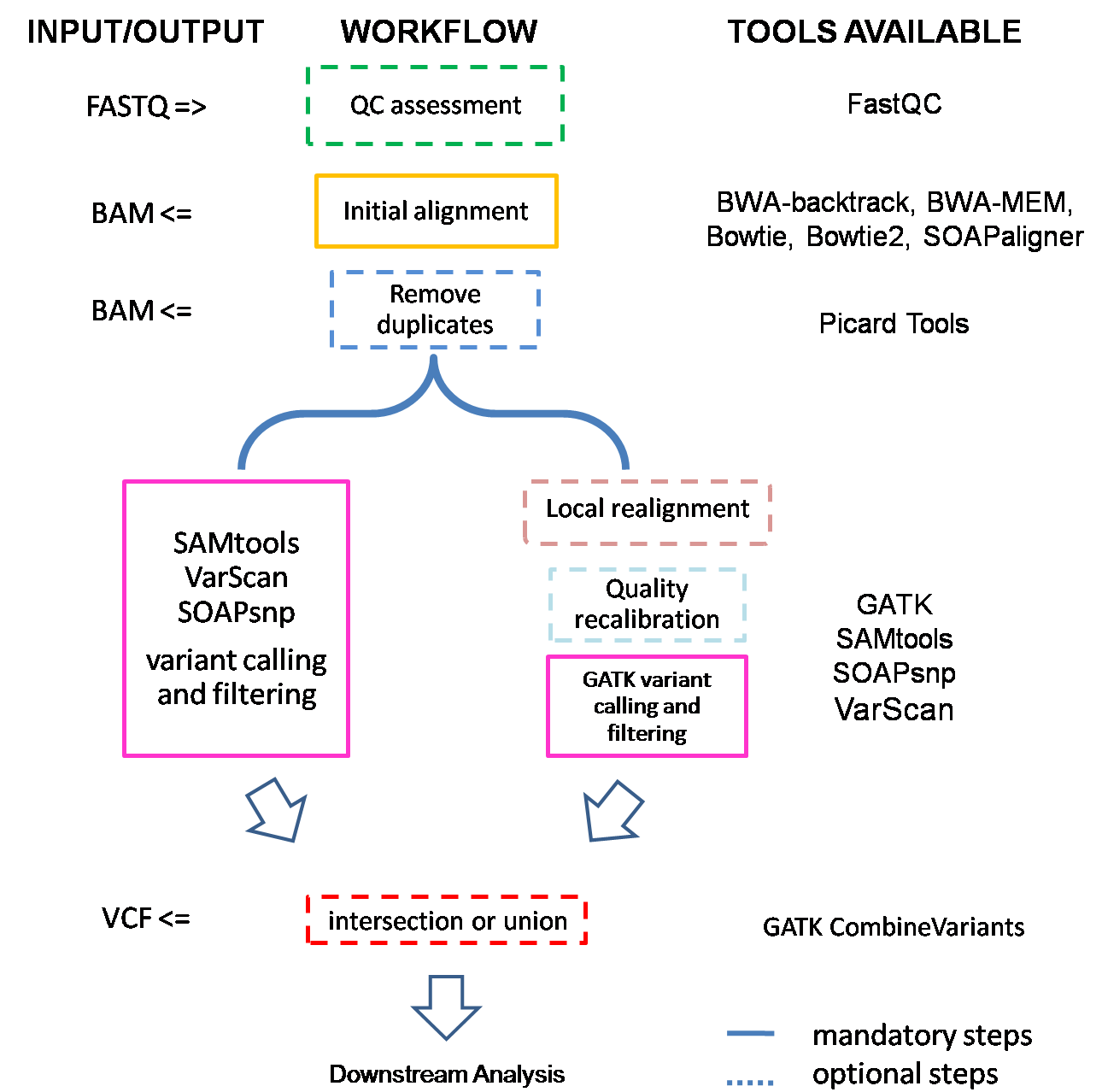
If input is FASTQ files, they will first be checked by FastQC to ensure sequencing quality. Then they are subjected to alignment., and we get BAM files after this step. Depending on the length of capture region, SeqMule will try to remove possible PCR and optical duplicates with PicardTools. Next one or more variant callers will be used to call variants. If we get multiple variant files, SeqMule will automatically extract consensus calls.
BAM input
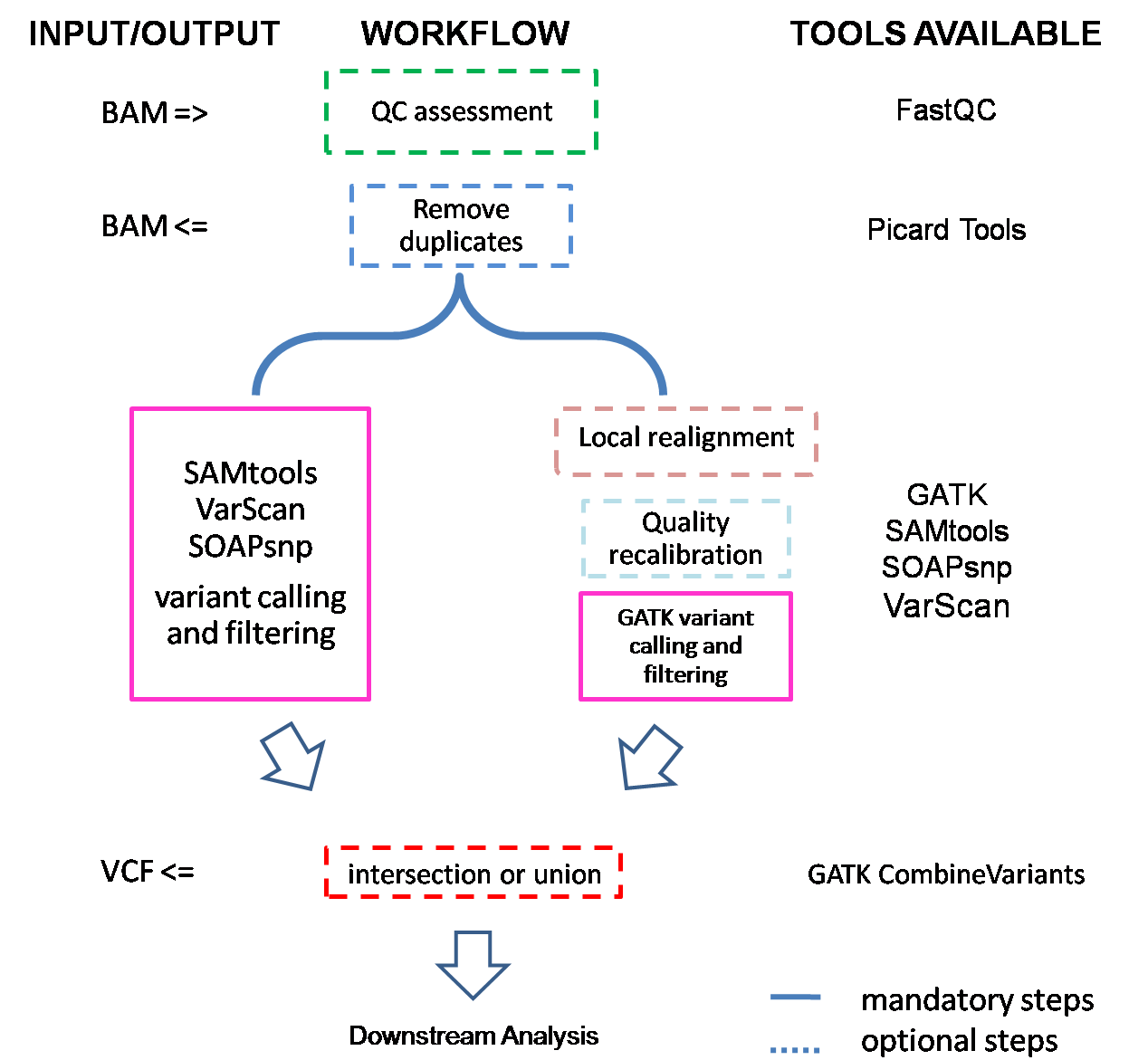
If input is BAM files, they will first be converted to FASTQ files, and then checked by FastQC to ensure sequencing quality. The rationale for this conversion is that it is hard to tell which reference genome was used for alignment. Then they are subjected to alignment., and we get BAM files after this step. Depending on the length of capture region, SeqMule will try to remove possible PCR and optical duplicates with PicardTools. Next one or more variant callers will be used to call variants. If we get multiple variant files, SeqMule will automatically extract consensus calls.