father
Input Quality Assessment
father.0.0_phred33
father.0.1_phred33
father.1.0_phred33
father.1.1_phred33
Statistics
| source: father_result/father_bwamem.merge.bam | |
| QC-passed reads | 3992668 |
| QC-failed-reads | 0 |
| Duplicates | 0 |
| Mapped reads | 3575179(89.54%) |
| Paired reads | 3992668 |
| Read1 | 1996332 |
| Read2 | 1996336 |
| Properly paired | 3914772(98.05%) |
| Reads with itself and mate mapped | 3574162 |
| Singletons | 1017(0.03%) |
| Reads with mate mapped to a different chromosome | 21721 |
| Reads with mate mapped to a different chr (mapQ at least 5) | 21721 |
| source: father_result/father_bwamem.merge.bam | |
| total length (defined by /auto/rcf-proj/kw/yunfeigu/Downloads/SeqMule/misc/hg19_exome.bed) | 46.21Mb |
| Fraction of reads mapped to target region | 53.28% |
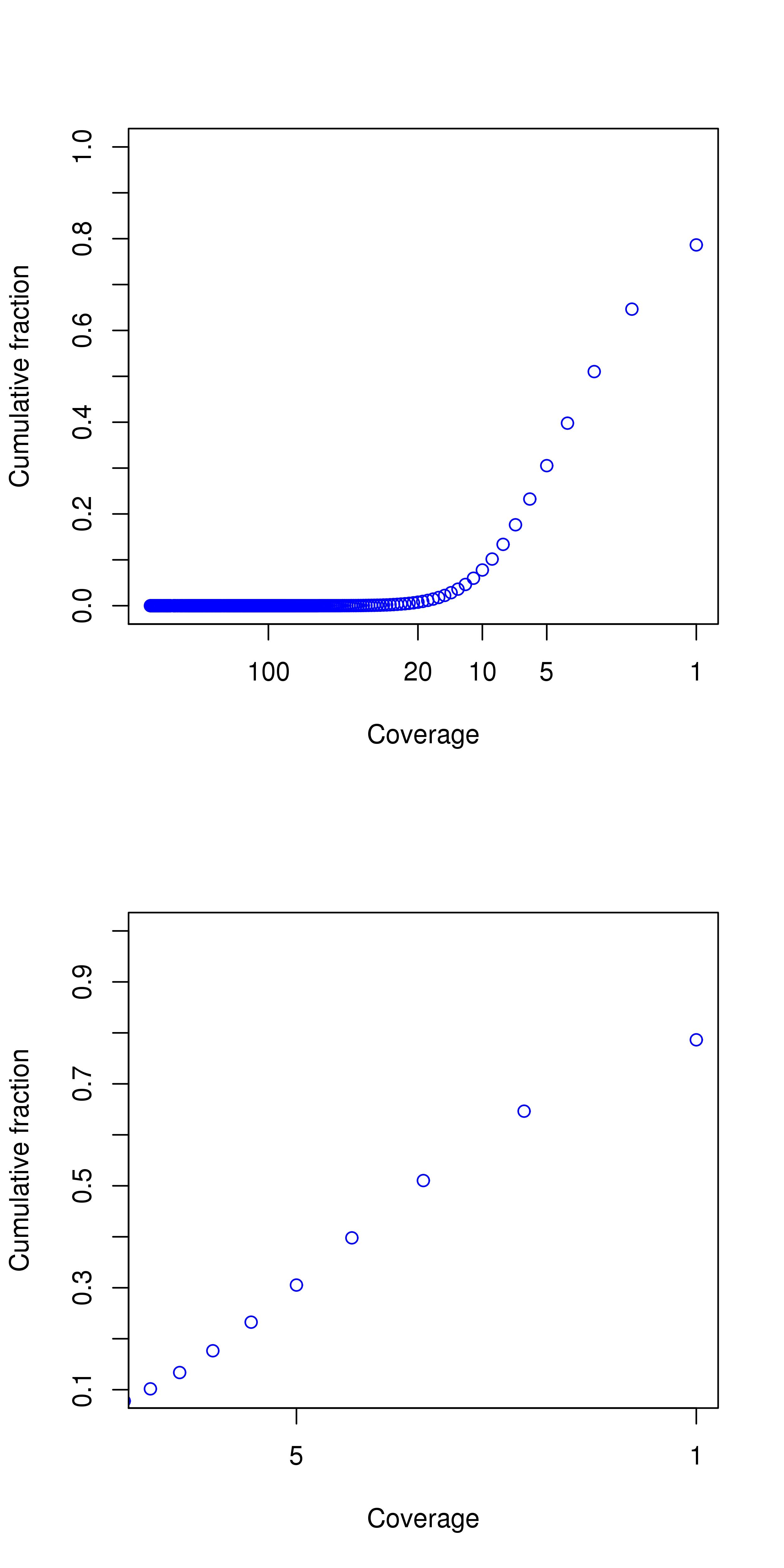
| Average coverage in target region | 3.66 |
| Percentage above 30 | 0.12% |
| Percentage above 20 | 0.77% |
| Percentage above 10 | 7.78% |
| Percentage above 5 | 30.53% |
| source: father_result/father_bwamem.merge.0_samtools.extract.vcf | |
| Sample | father |
| Number of variants | 3269 |
| Number of SNVs | 3203 |
| Number of indels | 66 |
| Transitions | 2196 |
| Transversions | 1007 |
| Ti/Tv Ratio | 2.18 |
| Ref/Alt heterozygotes | 2389 |
| Homozygotes | 880 |
Coverage Plot