son
Input Quality Assessment
son.1_phred33
son.2_phred33
Statistics
| source: son_result/son_bwamem.sort.rmdup.readfiltered.bam | |
| QC-passed reads | 138779950 |
| QC-failed-reads | 0 |
| Duplicates | 12571793 |
| Mapped reads | 124428082(89.66%) |
| Paired reads | 138779950 |
| Read1 | 69389975 |
| Read2 | 69389975 |
| Properly paired | 135643864(97.74%) |
| Reads with itself and mate mapped | 124356940 |
| Singletons | 71142(0.05%) |
| Reads with mate mapped to a different chromosome | 839981 |
| Reads with mate mapped to a different chr (mapQ at least 5) | 839981 |
| source: son_result/son_bwamem.sort.rmdup.readfiltered.bam | |
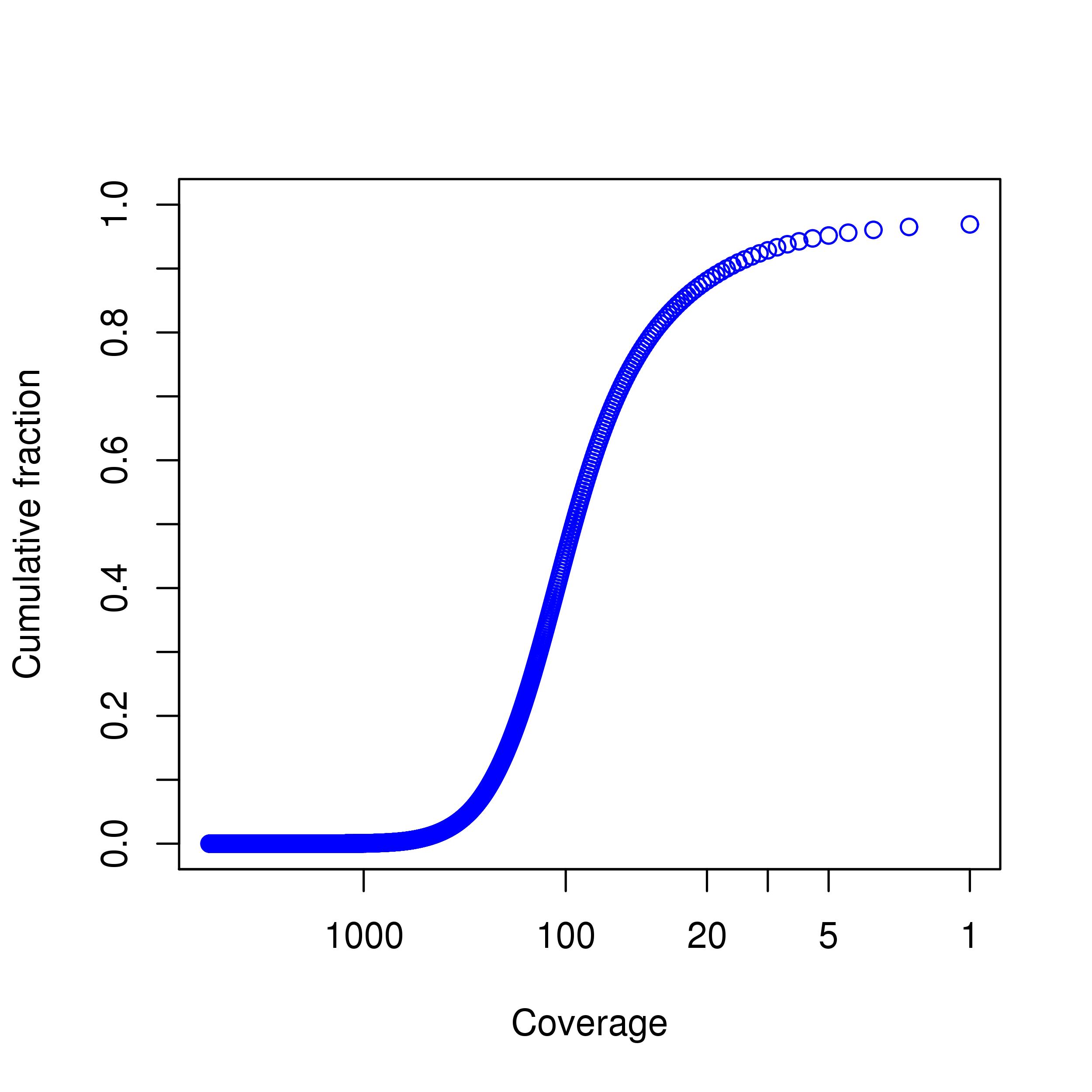
| total length (defined by /home/yunfeiguo/projects/seqmule_parallel/bin/secondary/../../misc/hg19_exome.bed) | 46.21Mb |
| Fraction of reads mapped to target region | 52.49% |
| Average coverage in target region | 113.44 |
| Percentage above 30 | 83.35% |
| Percentage above 20 | 88.11% |
| Percentage above 10 | 92.85% |
| Percentage above 5 | 95.17% |
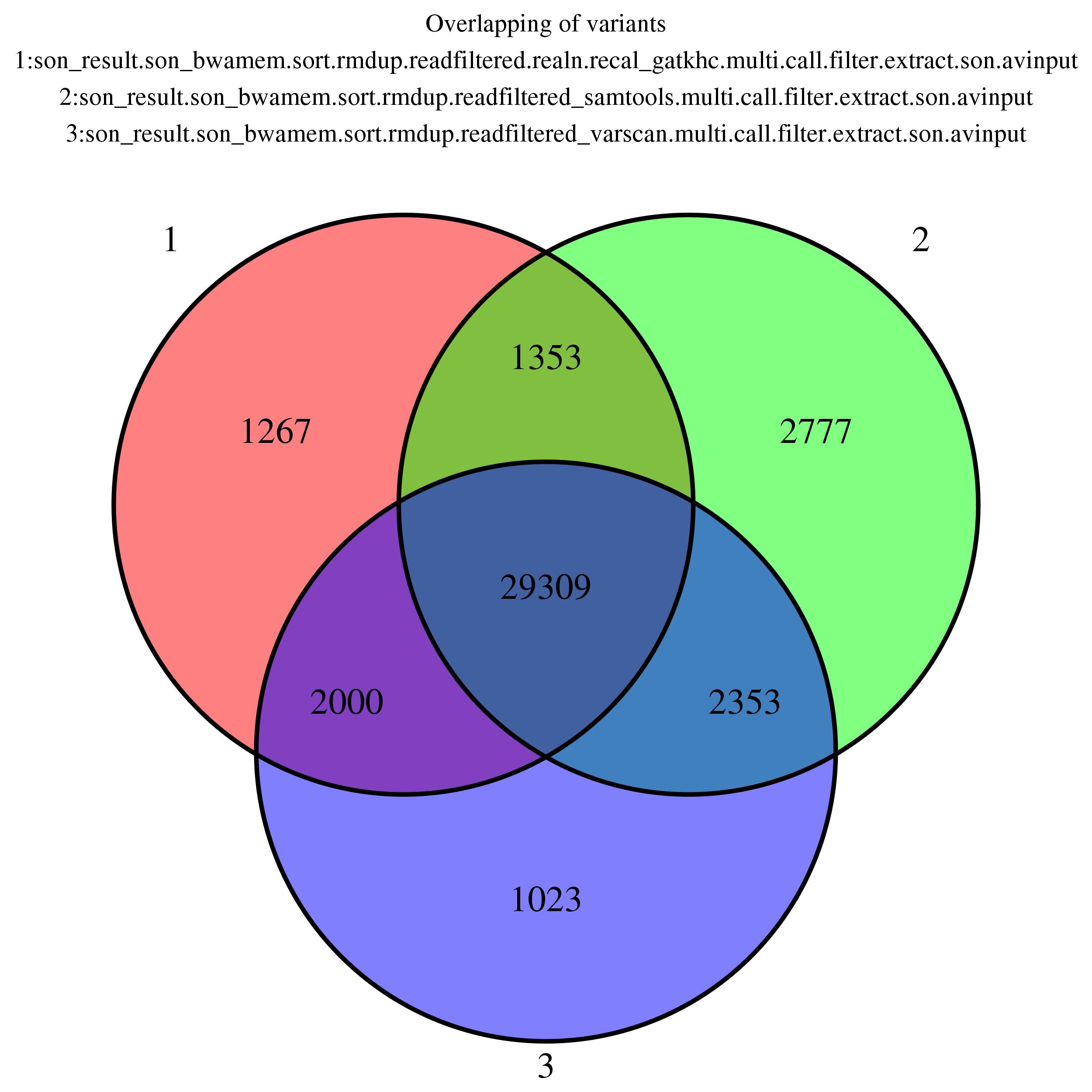
| source: son_result/son.extract_consensus.vcf | |
| Sample | son |
| Number of variants | 31897 |
| Number of SNPs | 30157 |
| Number of indels | 1740 |
| Transitions | 22306 |
| Transversions | 7863 |
| Ti/Tv Ratio | 2.84 |
| Total heterozygotes | 19658 |
| Ref/Alt heterozygotes | 19611 |
| Alt/Alt heterozygotes | 47 |
| Homozygotes | 12239 |
Venn Diagram
Showing overlapping of up to 5 variant output
Coverage Plot